
Cystic Fibrosis Challenges Proper Digestion and Nutrient Absorption
Written by |
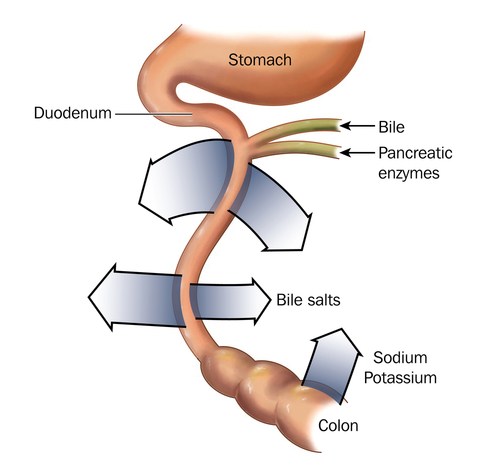



Patients with cystic fibrosis are all too familiar with the fact that a single mutation — in particular, a mutation in the gene coding for the CFTR protein — can wreak havoc on an individual’s health. Those afflicted with cystic fibrosis commonly face a number of problems in their digestive systems, causing poor digestion and inadequate nutrient intake.
A concise review concerning the multiple problems associated with cystic fibrosis was published online,, ahead of print in the journal Digestive and Liver Disease by a duo from Griffith University and Australian Catholic University, both in Brisbane, Australia. Within the article, authors Drs. Li Li and Shawn Somerset highlight the reasons for faulty digestion and absorption, as well as potential solutions to enhance nutrient uptake for energy production.
The most prominent problem with digestion is insufficient pancreatic enzyme activity. A normal pancreas is decorated with CFTR, but a deficiency of functional CFTR in cystic fibrosis leads to an altered pH and poor chloride ion secretion. As a result, cystic fibrosis patients suffer from exocrine pancreatic insufficiency, which leads to maldigestion and malabsorption.
To combat pancreatic insufficiency, a number of patients choose to use pancreatic enzyme replacement therapy, which is a pill containing a formulation of the enzymes lipase, protease, and amylase. These are known as pancreatic enzyme products, and the FDA has approved a handful to help aid food digestion. Yet without more approved formulations, pancreatic enzyme products can be costly, and there is no guarantee they will improve digestion and absorption. In fact, the enzymes can be compromised by an incorrect gastric pH through either deactivation or inactivation.
Adding to pancreatic insufficiency is insufficient bile activity. The altered pH of the intestine can precipitate bile and disallow lipid absorption. Without bile activity, not only are lipids not absorbed, but also lipid-soluble vitamins are not absorbed.
[adrotate group=”1″]
To combat the problem of bile inactivity, researchers have investigated gastric lipase to digest lipids. Unlike pancreatic enzyme products, gastric lipase has a high activity in acidic pH. In fact, gastric lipase takes care of 90% of lipid digestion in the upper small intestine in cystic fibrosis patients with pancreatic insufficiency.
Overall, large gains have been made in improving treatment of digestive issues in patients with cystic fibrosis. On the horizon of treatments is n-3 polyunsaturated fatty acid (PUFA) supplementation. PUFA is anti-inflammatory and anti-bacterial, allowing it to address the issues of inflammation and small intestine bacterial overgrowth. Additionally, identifying cystic fibrosis biomarkers–or abnormalities common to patients with cystic fibrosis–may help properly diagnose patients and even determine the response of patients to certain treatments. As a result, this growing body of research and drug development for treating the digestive and nutrient absorption issues associated with Cystic Fibrosis constitutes a much-needed complement to recent therapeutic breakthroughs for treating CF-related lung infections and risk factors.




Fran
You are so awesome! I don't believe I've truly read through
something like this before. So nice to discover another person with some genuine thoughts
on this issue. Seriously.. thank you for starting this up.
This site is something that is needed on the internet, someone with a bit of originality!