
 Fact-checked by
Fact-checked by  Discussion
Discussion
Cystic fibrosis overview
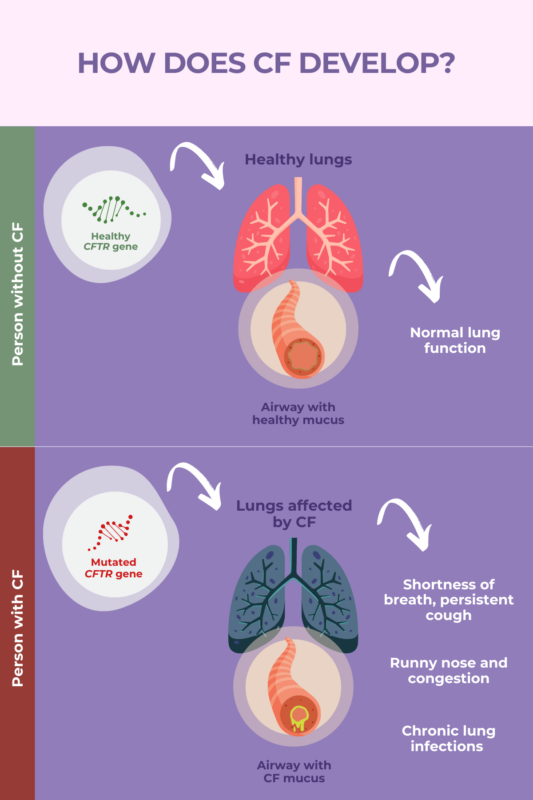
Cystic fibrosis, or CF, is a genetic disease caused by mutations inherited from an individual’s biological parents. As a result of these mutations, the body produces an abnormally thick and sticky form of mucus.
Normal mucus is mostly water, and it helps provide protection and lubrication to the body’s tissues. In CF, the sticky and thick mucus builds up in various organs, including the lungs, pancreas, liver, and intestines. This buildup of mucus is responsible for most symptoms of CF, such as lung and digestive problems.
The first formal description of the disease in modern medical literature was published in 1938 by American pathologist Dorothy Andersen. She described fluid-filled lumps (cysts) and scar tissue (fibrosis) in the pancreas, which is how CF got its name.
For most of human history, the majority of people born with CF have not survived past childhood. However, many treatments are now available to help ease cystic fibrosis symptoms and address disease complications. That has led to dramatically improved outcomes for patients — nowadays, the average life expectancy for someone born with CF is in the 60s, and that age is continuing to increase as treatments get better and better.
Causes
The causes of cystic fibrosis are mutations in the CFTR gene — short for cystic fibrosis transmembrane conductance regulator. This gene provides cells with instructions for making a protein, also called CFTR, that normally functions like a gated channel on the surface of certain cells. By opening or closing the this so-called gate, this protein helps regulate the flow of chloride ions, a type of negatively charged salt particle.
In cystic fibrosis, mutations cause the CFTR protein to be dysfunctional or missing. As a result, cells cannot properly regulate the flow of salt and water, leading to the production of the thick and sticky mucus that characterizes the disease.
More than 2,500 different mutations in the CFTR gene have been reported. These mutations may be categorized into various types based on the effect on the CFTR protein:
- Class 1, which are protein production mutations, result in little to no CFTR protein being made.
- Class 2, protein processing mutations, result in an unstable protein that does not reach the cell surface.
- Class 3, which are gating mutations, cause the gate-like CFTR protein to become stuck in a closed type of position.
- Class 4, conduction mutations, change the shape of the channel-like protein so it’s harder for chloride to pass through.
- Class 5, which are insufficient protein mutations, allow the production of a functional CFTR protein, but levels much lower than normal are found at the cell membrane.
How is CF inherited?
Everyone inherits two copies of the CFTR gene, one from each biological parent. Cystic fibrosis is a recessive condition, which means that a person will only develop the disease if both copies of the gene are mutated. That means both parents must pass along a mutated gene copy to any offspring for that child to have CF.
A person with one mutated copy of the CFTR gene and one healthy copy is known as a carrier. Carriers do not develop CF symptoms, but they may pass the disease-causing mutation on to their biological children. If two carriers have biological children, there is a:
- 1 in 4 or 25% chance the child will have CF
- 1 in 2 or 50% chance the child will be a carrier but not have CF
- 1 in 4 or 25% chance the child will not have CF and will not be a carrier.
Symptoms and complications
Cystic fibrosis symptoms and complications can vary substantially from person to person; any one person with the disease may not experience every symptom associated with CF. Symptoms also can ease or worsen over time, and some symptoms may not become apparent until after childhood.
The first early signs of cystic fibrosis usually develop soon after birth. Babies with CF commonly experience symptoms such as:
- intestinal blockage
- greasy, foul-smelling stools
- difficulty gaining weight
- coughing and wheezing
- lung infections
- salty-tasting skin.
The thick mucus that characterizes CF builds up in the lungs, clogging the airways and providing a fertile breeding ground for infectious bacteria. This can lead to respiratory symptoms such as:
- wheezing, coughing, and shortness of breath
- frequent or chronic lung infections
- runny nose and congestion
- noncancerous growths, called polyps, in the nose.
Mucus also builds up in digestive organs, including the pancreas and intestines, which can interfere with normal digestion. Symptoms resulting from this buildup include:
- poor growth and difficulty putting on weight
- frequent stools that are unusually bulky, greasy, and smelly
- constipation, bloating, and flatulence
- stomach pain and heartburn
- intestinal blockages
- CF-related diabetes.
CF also can cause complications related to the reproductive tract, many of which particularly impact adolescents and adults. Among these symptoms are:
- complications with fertility
- delayed puberty
- irregular or absent menstrual periods
- frequent yeast infections
- urinary incontinence.
As CF worsens over time, patients may be at risk of other complications, such as:
- enlarged heart
- collapsed lungs
- damage to the lungs’ airways, known as bronchiectasis
- osteopenia or osteoporosis, where the bones are weaker than normal
- nutritional deficiencies
- liver disease
- colorectal cancer.
Diagnosis
Nowadays, most people with CF are diagnosed early in life through newborn screening programs, which test for the disease in every child at birth. Newborn screening programs for CF have been implemented in all 50 U.S. states and in many other countries around the world.
IRT test
Newborn screening usually involves taking a drop of blood and testing for immunoreactive trypsinogen or IRT, a protein made by the pancreas that builds up to high levels in the blood of CF patients.
However, other conditions also may cause elevated IRT levels, so additional testing is needed to confirm the diagnosis after a positive result from newborn screening.
Sweat test
The gold standard for confirming a cystic fibrosis diagnosis is the sweat test, which involves measuring levels of chloride in patients’ sweat. Defects in CFTR protein function lead to abnormally high levels of chloride in sweat.
The Cystic Fibrosis Foundation recommends that babies who are positive for newborn screening via the IRT test should get the sweat test by the age of 1 month, at the latest. Babies must be old enough to produce sufficient sweat for the test to be done, meaning it typically is not done immediately after birth.
Genetic testing
Genetic testing is used to identify the specific disease-causing mutations in the CFTR gene. This testing may be used to help confirm the diagnosis and determine eligibility for certain CF treatments.
Such testing also may be used to identify a disease carrier or to check for cystic fibrosis in a developing fetus during pregnancy. Identifying carriers can allow prospective parents to consider reproductive options.
Positive results from genetic testing are highly accurate. However, there are so many CFTR mutations and most tests only look for the most common ones. Thus, false-negative results may occur, and are more common in nonwhite populations.
Treatment
While there is no cure for CF, a wide array of therapies are available that can help ease symptoms and slow disease progression for people living with cystic fibrosis.
CFTR modulators are a recently developed class of therapies that can boost the functionality of the CFTR protein in people with specific disease-causing mutations. By improving the protein’s function, these therapies can allow more normal mucus production, which can ease symptoms and slow disease progression.
There are four modulator therapies now approved in the U.S., three of which containing so-called CFTR correctors and all containing the CFTR potentiator ivacaftor. All are marketed by Vertex Pharmaceuticals.
- Kalydeco (ivacaftor)
- Orkambi (ivacaftor/lumacaftor)
- Symdeko (ivacaftor/tezacaftor)
- Trikafta (ivacaftor/tezacaftor/elexacaftor)
In addition to modulators that directly target the underlying causes of cystic fibrosis, a variety of other medications are commonly used to help manage the disease. These include:
- antibiotics to treat bacterial infections
- inhaled therapies like bronchodilators or mucus thinners to help ease respiratory symptoms and clear mucus
- pancreatic enzyme replacement therapy, called PERT, to help manage digestive health and ensure adequate nutrition.
Apart from medication, healthy lifestyle habits — especially eating a balanced diet and getting physical exercise — are a key part of managing the disease for most patients, and can help to better control symptoms. Patients are encouraged to avoid smoking and exposure to secondhand smoke, be active, and eat high-energy foods.
Many people with CF also benefit from nonmedication therapies such as mucus-clearing techniques, physical therapy, and supplemental oxygen. Such options can be discussed with the patient’s healthcare team.
Prognosis
Over the past century, as scientific understanding has deepened and care has advanced, the prognosis for people born with CF has improved dramatically. Although a cure has not been discovered, modern treatment options have allowed much better quality of life and a more positive prognosis for most cystic fibrosis patients.
In the 1950s, the majority of people with CF did not survive past the first few years of life. By the late 1970s, the estimated median survival for people with CF in the U.S. was 11 years, and in the decades since, life expectancy has continued to climb — by the early 2010s, when the first CFTR modulators started getting approved, life expectancy for someone born with cystic fibrosis was 40 years.
Based on data from the Cystic Fibrosis Foundation’s patient registry, the average life expectancy for people with CF born between 2018 and 2022 is estimated at 56 years. Based on the same data, half of the people with CF born today are expected to live into their 60s, if not longer.
While the prognosis has improved substantially, CF remains a chronic illness, and patients will generally require lifelong medical care to help them stay healthy, manage disease complications, and maximize their quality of life. It’s also important to note that, while overall prognostic outcomes have improved drastically, the individual prognosis will vary from patient to patient, depending on a person’s specific situation.
Finding new therapies that can offer benefit to patients not eligible for available CFTR modulator treatments has emerged as a big priority for the CF research community in recent years, and several potential candidates are in development.
Cystic Fibrosis News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Your CF Community

Visit the Cystic Fibrosis News Today forums to connect with others in the CF community.
FAQs about cystic fibrosis
Category:
Overview
According to the Cystic Fibrosis Foundation, there are nearly 40,000 people living with cystic fibrosis in the U.S., and more than 100,000 people living with the disease worldwide. Cystic fibrosis can affect people of every race and ethnic group.
Category:
Overview
Cystic fibrosis is characterized by the production of abnormally thick, sticky mucus. This mucus builds up in the body’s organs, particularly the lungs and digestive system, and is mainly responsible for driving disease symptoms. The buildup of thick mucus can cause clogs that interfere with organ function, and it sets the stage for infections and inflammation that cause progressive damage over time.
Category:
Overview
Everyone inherits two copies of the CFTR gene, mutations in which underlie cystic fibrosis (CF), one from each biological parent. CF is inherited in an autosomal recessive manner, which means the disease will only develop if there is a mutation in both copies of this gene.
Category:
Overview
Cystic fibrosis is a genetic disorder caused by mutations that are inherited from a person’s biological parents. It is not contagious. However, people with CF may be at increased risk from infections, so patients often have to take extra precautions to avoid other diseases that are contagious.
Category:
Overview
Cystic fibrosis (CF) is a recessive condition, meaning the disease will only develop if an individual carries mutations in both copies of the CFTR gene, one inherited from each biological parent. A person with one mutated copy of the CFTR gene alongside a second healthy copy will not develop CF but may pass the disease-causing mutation on to any biological children. Such individuals are called carriers.
Related Articles
-
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion