
 Fact-checked by
Fact-checked by  Discussion
Discussion
Types of CFTR mutations
Cystic fibrosis (CF) is a progressive genetic disorder caused by a mutation in the CFTR gene, which encodes a protein of the same name. To date, more than 2,500 different mutations in the CFTR gene have been described. Patients can have more than one CFTR gene mutation.
The specific type of disease-causing mutation may affect CF severity. Because different mutations cause varying changes to the protein, they also may affect what treatment options are available for a given individual. For example, CFTR modulators are a type of medication that can address defects in the CFTR protein caused by some specific types of mutations but not others.
The Cystic Fibrosis Foundation offers a mutation analysis program where people who have been diagnosed with definitive or probable CF can access genetic testing to determine what type of CF-causing mutation they have.
How are different types of CFTR mutations categorized?
All CF-causing mutations lead to reduced or no function of the CFTR protein. Normally, once the CFTR protein is made, it is shuttled to the cell membrane where it acts like a gate, helping to regulate the flow of chloride ions in and out of cells. The proper regulation of this flow is important for the production of mucus; when it’s impaired, mucus can become abnormally thick and sticky. The buildup of thick mucus in the body’s organs is mainly responsible for driving CF symptoms.
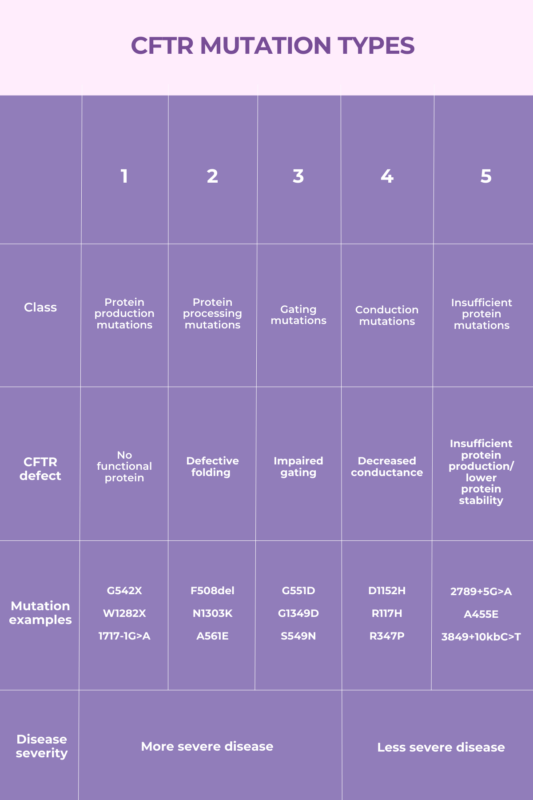
CF-causing mutations are generally categorized based on the specific effect they have on the CFTR protein. Over the years, different classification systems have been used. At present, CF-causing mutations are usually divided into five classes, based on their impact on protein production.
- Class 1 includes protein production mutations.
- Class 2 involves protein processing mutations.
- Class 3 consists of gating mutations.
- Class 4 involves conduction mutations.
- Class 5 includes insufficient protein mutations.
In general, mutations resulting in a more substantial defect in the protein are linked to more severe disease.
Protein production mutations
Protein production mutations, also called class 1 mutations, result in little to no CFTR protein being produced. This is due to problems that occur when the CFTR gene is assessed, or read, to make protein. According to a 2022 review study, mutations of this type are found in about 10% of CF patients.
There are two types of protein production mutations: nonsense mutations and splicing mutations.
Nonsense mutations
A nonsense mutation causes a type of stop signal to be present in the middle of a gene — sort of like placing a period in the middle of a sentence. As a result, when the cell’s protein-making machinery tries to read the genetic sequence, it ends up stopping too early. This leads to an unstable protein being produced, which gets rapidly degraded by cells. Common nonsense mutations that cause CF include:
- G542X
- W1282X
- R553X.
Splicing mutations
Like other protein-coding genes, the one for CFTR contains stretches of genetic code, known as exons, that contain instructions for making protein. There also are other sections, called introns, that are important for regulating gene activity but aren’t necessary for building the CFTR protein.
When the CFTR gene is read, the entire sequence gets copied into a temporary molecule, known as messenger RNA, and normally the parts that aren’t needed to make protein are removed. Splicing mutations cause problems with this process, resulting in incorrect instructions for intron removal, and ultimately preventing CFTR protein from being produced. Examples of splicing mutations include:
- 1717-1G>A
- 621+1G>T.
Because CFTR modulator therapies work by targeting the CFTR protein itself, these medications are not expected to provide any benefit for patients with mutations that prevent the protein from being made. With a few exceptions, most CF patients with splicing mutations are not eligible to receive CFTR modulators.
Protein processing mutations
The CFTR protein normally needs to fold into a specific three-dimensional shape to function properly. Class 2 or protein processing mutations result in abnormal folding of the CFTR protein. As a result, the protein is not stable, so it ends up getting destroyed before it’s able to get to the cell membrane and carry out its function.
Protein processing mutations are the most common type of CF-causing mutation — nearly 9 in 10 patients carry at least one copy of this type of mutation. A protein processing mutation called F508del is the most common CF-causing mutation. Other examples of protein processing mutations include:
- N1303K
- I507del
- A561E.
CFTR correctors — which include lumacaftor, elexacaftor, and tezacaftor — are a type of CFTR modulator therapy that can bind to the mutated CFTR protein and help it to fold correctly.
Three medications containing CFTR correctors are approved at present in the U.S. All three of these therapies are sold by Vertex Pharmaceuticals.
- Trikafta (elexacaftor/tezacaftor/ivacaftor) is approved to treat patients ages 6 and older who have at least one copy of the F508del mutation or another mutation that responds to therapy based on data from cell experiments.
- Orkambi (ivacaftor/lumacaftor) is approved for patients 1 and older with two copies of the F508del mutation.
- Symdeko (tezacaftor/ivacaftor) is approved for patients 6 and older who have one of more than 150 different mutations.
Gating mutations
The CFTR protein normally functions like a gated channel on the cell surface: the gate can open to allow chloride ions to flow through, then close to stop the flow. Gating mutations, also called class 3 mutations, cause the gate to become stuck closed, so the protein is no longer able to function.
About one of every 25 CF patients carries a gating mutation. Some common gating mutations include:
- G551D
- S549N
- G1349D.
CFTR potentiators are a class of medications that can help to prop open the gate in the CFTR protein, allowing the protein to function in people with gating mutations. The CFTR potentiator ivacaftor is sold by Vertex as Kalydeco, which is approved in the U.S. to treat people with CF caused by one of 97 different mutations. Ivacaftor also is contained in Trikafta, Orkambi, and Symdeko.
Conduction mutations
The CFTR protein normally functions like a channel, allowing chloride ions to flow through the protein in and out of the cell. Class 4 or conduction mutations disrupt the structure of this channel, making it harder for chloride to flow through — a bit like having a clogged pipe.
Conduction mutations are estimated to be present in less than 2% of CF patients, or about one of every 50. Examples of this mutation type include:
- D1152H
- R347P
- R117H.
This type of mutation may be treated with CFTR potentiators such as Kalydeco, which help to prop open the CFTR protein. Other CFTR modulator therapies such as Trikafta, which contain potentiators along CFTR correctors, also may benefit patients with these mutations.
Insufficient protein mutations
In class 5 mutations, a normal version of the CFTR protein is made, but the protein is present at levels much lower than normal, so there’s not enough of the protein to adequately do its job.
Some insufficient protein mutations result in less CFTR protein being made by the cell. In other mutations, the cell produces CFTR protein, but the protein is unstable on the cell’s surface, so it is rapidly degraded. In either case, the end result is that there is functional CFTR protein at the cell’s surface, but in lower than normal quantities that are insufficient.
Class 5 mutations are very rare. Examples include:
- 3849+10kbC>T
- 2789+5G>A
- A455E.
CFTR potentiators like Kalydeco, which can increase CFTR function by propping the gate-like protein open, may benefit CF patients with insufficient protein mutations. Trikafta, which contains CFTR potentiators and correctors, also may be of benefit to some patients with particular insufficient protein mutations.
Does CFTR mutation type affect life expectancy?
Generally, mutations that result in a more substantial defect in the CFTR protein are associated with more severe disease and, consequently, shorter life expectancy. As such, CF patients with protein production, protein processing, or gating mutations (classes 1-3) tend to have more severe disease than those with conduction or insufficient protein mutations (classes 4-5).
Because everyone inherits two copies of the CFTR gene — one from each biological parent — the combination also affects disease severity. For example, patients with class 1-3 mutations in both copies of the gene tend to have more severe disease than patients with a class 1-3 mutation in one copy and a class 4-5 mutation in the other.
It’s important to stress that these are general trends, and there can be a lot of variability from person to person in how CF manifests and progresses. Also, CFTR modulators and other treatments can profoundly alter outcomes, especially when initiated early.
Cystic Fibrosis News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Your CF Community

Visit the Cystic Fibrosis News Today forums to connect with others in the CF community.
FAQs about types of CFTR mutations
Category:
CFTR mutations
Some treatments for cystic fibrosis (CF) will only work in patients with specific disease-causing mutations in the CFTR gene. Most notably, a class of medications called CFTR modulators can work to improve the functionality of the CFTR protein in people with specific disease-causing mutations, but these therapies will not work for people with other mutation types.
Category:
CFTR mutations
According to the Johns Hopkins Cystic Fibrosis Center, more than 2,500 different mutations in the CFTR gene have been documented.
Category:
CFTR mutations
Mutations in the CFTR gene are classified based on their effect on the CFTR protein. A common modern classification system groups mutations into five classes: protein production mutations (class 1), protein processing mutations (class 2), gating mutations (class 3), conduction mutations (class 4), and insufficient protein mutations (class 5).
Category:
CFTR mutations
Mutations in the CFTR gene cause cystic fibrosis (CF). The CFTR gene provides instructions for making the CFTR protein, which is important for regulating the production of mucus in the body. Mutations in CFTR lead to the production of abnormally thick and sticky mucus which builds up in the body’s tissues, causing damage that drives most CF symptoms.
Category:
CFTR mutations
The most common mutation that causes cystic fibrosis (CF) is called F508del. This mutation is present in up to about 90% of CF patients, though it is more common in white populations than in other racial and ethnic groups.
Related Articles
-
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion