
Translarna Shows Potential to Treat CF and Other Nonsense Mutation Disorders in Study
Written by |
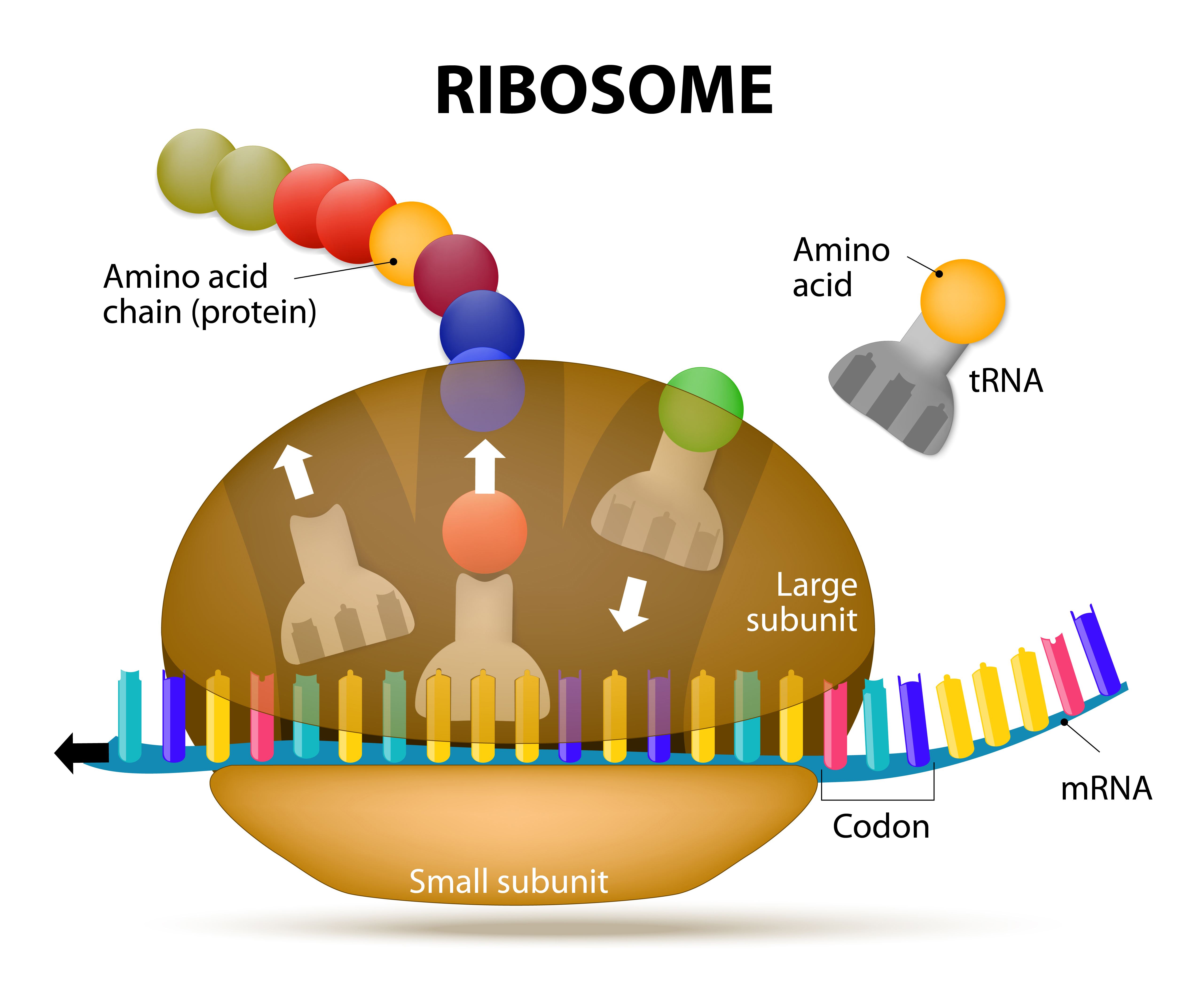



The experimental drug Translarna (ataluren) can make the cellular protein-making machinery run the so-called “stop sign” mutation in the cystic fibrosis (CF)-causing CFTR gene, replacing it with amino acids (the protein building blocks) similar enough to allow the protein to do its work.
Translarna is currently in Phase 3 clinical trials for CF patients carrying such stop signs, known as nonsense mutations, in their CFTR gene.
The study, “Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression,” was published in the journal Proceedings of the National Academy of Sciences (PNAS).
Gene mutations that substitute the code for an amino acid with another code — representing a stop in a sequence — lead to the production of faulty proteins, which can cause severe diseases. If it were possible to force the cellular machinery to ignore such stop signals, the very cause of diseases like CF could be treated.
Translarna is an attempt to do just that. The drug was developed by PTC Therapeutics, a company founded by Allan Jacobson, PhD, the study’s senior author at the University of Massachusetts Medical School, and David Bedwell, PhD, from the University of Alabama at Birmingham.
Although the drug has reached Phase 3 studies (NCT02107859 and the extension trial NCT02456103) in patients with cystic fibrosis, up to now, researchers have not been sure as to which amino acid is being inserted instead of the stop signal.
If the inserted part differs significantly from the natural one, it could change the properties of the protein, essentially replacing one problem with another. But, if the drug can force the machinery to insert a similar building block, the protein would be more likely to work properly.
To find out, researchers used human and yeast cells, grown in the lab, and created stop mutations in six different genes. An analysis of the resulting proteins confirmed that Translarna triggered insertions of similar amino acids, acting on what researchers call near-cognate tRNAs. tRNAs are the molecules bringing amino acids, corresponding to a particular code, to be added to the protein that is being made.
The study also found that the resulting proteins had the correct length, meaning that Translarna recognized the “real” stop sign and produced a protein neither too long nor too short. It did not disrupt other aspects of protein synthesis.
“These results further support our clinical findings demonstrating the production of full-length functional protein in nonsense mutation Duchenne muscular dystrophy and cystic fibrosis,” Stuart Peltz, PhD, co-founder and chief executive officer of PTC Therapeutics, said in a University of Alabama News report. “Given this mechanism, ataluren offers the potential for a new therapeutic approach for multiple nonsense mutation genetic disorders by targeting the underlying cause of the disease.”
Researchers also specifically looked at the amino acids inserted instead of the stop sign in the CFTR gene holding the G542X nonsense mutation, the most common nonsense mutation in this gene. They noted that three different amino acids were used. The resulting protein was functional, meaning that it ended up in the cell membrane and did its work as an ion channel. The study showed that the proteins produced mimicked the natural CFTR protein to various degrees, depending on which amino acid was present.
Finally, the study demonstrated that the antibiotic tobramycin, often used in CF patients, prevented Translarna from working. The helps to explain data from the current clinical trial, showing that the drug only benefitted patients who were not treated with tobramycin.
“Our new data is scientifically important because ataluren restores activity to genes inactivated by nonsense mutations, and as a result, it has the potential to do so much for a large number of very complex genetic disorders,” Jacobson concluded.



