
Suppressing Function of Chloride Channel TMEM16A Eyed as Potential Therapeutic Approach



Researchers have reported the case of two siblings with impaired CFTR function due to a deficiency in another chloride channel, called TMEM16A, that do not present symptoms associated with cystic fibrosis (CF).
While there is evidence supporting either the activation or the suppression of TMEM16A activity as a potential treatment of CF, these findings support the latter. Clinical trials evaluating the therapeutic benefit of blocking TMEM16A function in CF patients are on the horizon, researchers believe.
The study, “TMEM16A deficiency: a potentially fatal neonatal disease resulting from impaired chloride currents,” was published in the Journal of Medical Genetics.
CF is caused by a deficiency in CFTR, a cell membrane channel involved in water balance by regulating negatively charged ions, specifically chloride ions, in and out of cells. CFTR deficiency results in the production and accumulation of thick, sticky secretions and mucus in the lungs, digestive tract, and reproductive system.
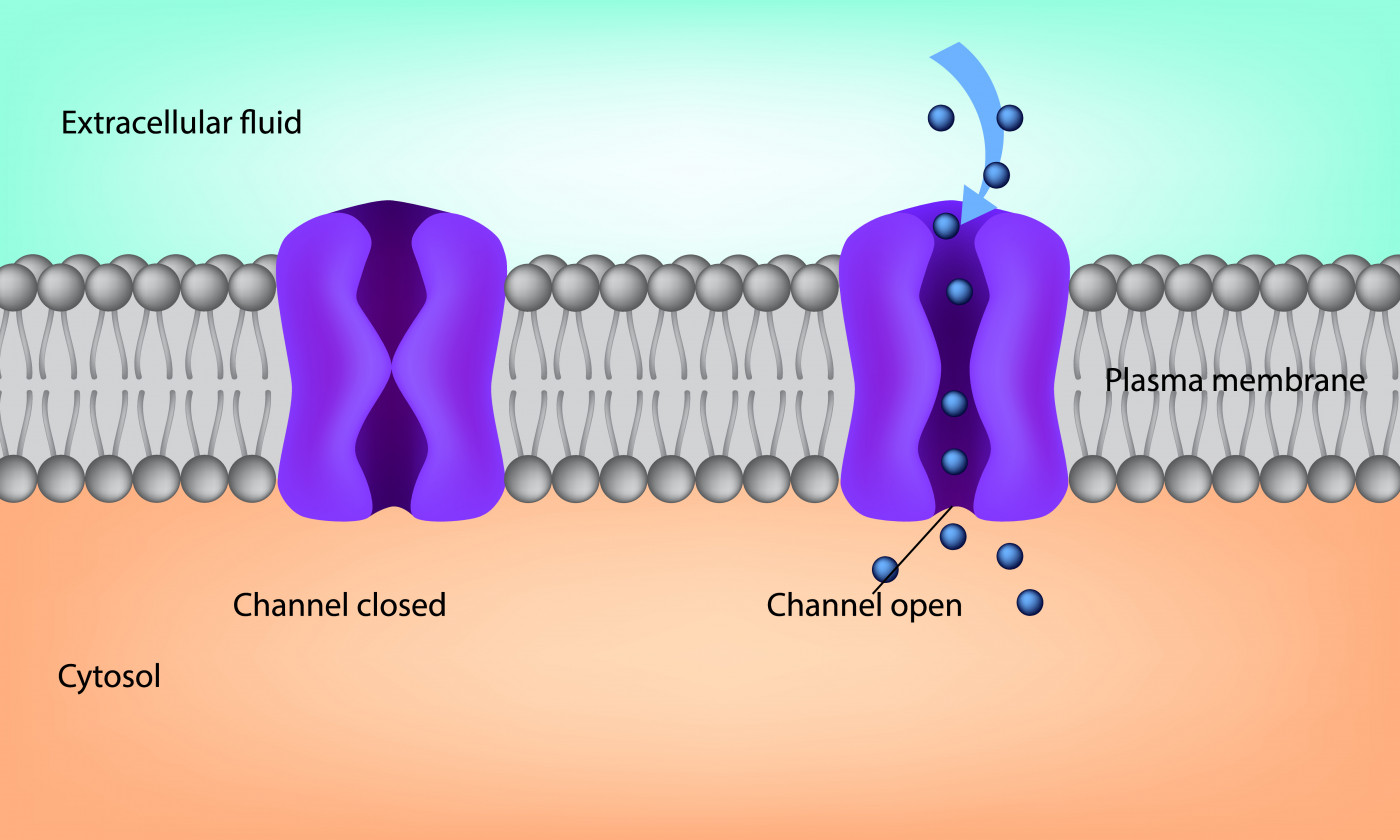
TMEM16A is another chloride channel generally located in the same tissues as CFTR. In the lungs, it plays a major role in the natural production of mucus and is indispensable for CFTR function. However, it also contributes to excessive mucus production in airway inflammation and in CF.
TMEM16A has been suggested as a potential therapeutic target in CF, but conflicting evidence exists on which approach — activation or suppression of TMEM16A — will result in greater benefits for people with CF.
Now, the absence of CF-associated symptoms in two siblings with a previously unknown severe disease caused by TMEM16A deficiency suggested that blocking TMEM16A activity indeed may be an effective therapeutic approach for CF.
Researchers in Germany described the case of two infants, born from blood-related parents of Kurdish–Syrian origin, with impaired intestinal movements, swollen intestinal loops, presence of gas within the intestinal wall, malformations, and developmental problems.
Genetic analysis of the whole family — the parents, the two affected siblings, and an unaffected sister — confirmed the presence of a mutation in ANO1, the gene containing the instructions to produce TMEM16A protein.
The affected siblings carried the mutation in both copies of the ANO1 gene, while their parents and unaffected sister had one mutated ANO1 gene copy and one healthy copy.
Further tests revealed that this mutation led to the production of a shorter, non-functional TMEM16A protein, which resulted in impaired chloride balance. Not surprisingly, TMEM16A deficiency also affected the location and function of CFTR in cells from the respiratory tract.
However, these infants showed no typical signs and symptoms of CF and no issues in the lungs or pancreas.
“We were astonished that children with TMEM16A deficiency don’t have any respiratory symptoms at all. A loss of CFTR function due to lack of TMEM16A does not lead to clinical symptoms of cystic fibrosis in these kids,” Julien Park, MD, the study’s first author and researcher at the Münster University Hospital, in Germany, said in a press release.
The siblings’ clinical features were similar to those previously reported in mice lacking TMEM16A and CFTR, the team noted.
“The absence of [respiratory symptoms] raises questions about the current understanding of CF [underlying mechanisms] and indicates that [suppressing] TMEM16A function might be a promising therapeutic approach for CF,” the researchers wrote.
The team also noted that previous studies have shown that TMEM16A suppression promotes a significant drop in mucus production, which may prevent mucus buildup and improve mucus clearance in people with CF and other inflammatory lung diseases.
Karl Kunzelmann, MD, PhD, a study’s senior author at the University of Regensburg, Germany, said the team is already “planning clinical trials to evaluate a treatment of cystic fibrosis with TMEM16A inhibitors.”
Already available TMEM16A suppressors include niclosamide and idebenone.



