
UNC Researchers Find New CF Drugs Unsuspectedly Counteract One Another’s Effectiveness




 Findings of new study led by Martina Gentzsch, PhD, at the University of North Carolina School of Medicine and the UNC Marsico Lung Institute in Chapel Hill, N.C. could help drug developers improve compounds designed to correct CFTR proteins in cystic fibrosis (CF) patients. In lab experiments using tissue samples cultured from cystic fibrosis patients, UNC Dr. Gentzsch and her team of scientists have demonstrated that a new CF drug counteracts the intended beneficial molecular effect of another CF drug.
Findings of new study led by Martina Gentzsch, PhD, at the University of North Carolina School of Medicine and the UNC Marsico Lung Institute in Chapel Hill, N.C. could help drug developers improve compounds designed to correct CFTR proteins in cystic fibrosis (CF) patients. In lab experiments using tissue samples cultured from cystic fibrosis patients, UNC Dr. Gentzsch and her team of scientists have demonstrated that a new CF drug counteracts the intended beneficial molecular effect of another CF drug.
The finding, published in the journal Science Translational Medicine, shows how a mutant CFTR protein becomes unstable and loses its ability to function properly when in the presence of the two drugs. The research offers several insights into how novel CF pharmacotherapies could be improved.
The Science Translational Medicine study, entitled “Potentiator ivacaftor abrogates pharmacological correction of F508 CFTR in cystic fibrosis,” (Sci Transl Med 23 July 2014: Vol. 6, Issue 246, p. 246ra96 DOI: 10.1126/scitranslmed.3008680) is coauthored by Dr. Martina Gentzsch; Research Assistants Deborah M. Cholon, Nancy L. Quinney and M. Leslie Fulcher; UNC postdoctoral fellow Jhuma Das, PhD; Charles R. Esther Jr. MD, PhD; Nikolay Dokholyan, PhD, the Michael Hooker Distinguished Professor of Biochemistry and Biophysics; Scott H. Randell, PhD, Associate Professor of Cell Biology and Physiology; and Richard Boucher, MD, the Director of the UNC Marsico Lung Institute and the James C. Moeser Eminent Distinguished Professor of Medicine, and Richard C. Boucher; variously of the University of North Carolina Marsico Lung Institute/Cystic Fibrosis Research Center; Division of Pediatric Pulmonology, Department of Pediatrics; Department of Biochemistry and Biophysics; Department of Medicine; and Department of Cell Biology.
[adrotate group=”1″]
Cystic fibrosis results from defective epithelial salt and fluid transport that is caused by mutations in CFTR (cystic fibrosis transmembrane conductance regulator). People with cystic fibrosis have two faulty copies of the CFTR gene. In one type of mutation, patients don’t have enough CFTR proteins that transit normally to the cell surface. In other patients, mutant CFTR proteins that do transit to the cell surface don’t function properly. Both genetic mutations lead to poor mucus clearance from lungs, chronic lung infections, bouts of inflammation, and a decreased ability to breathe properly, among other symptoms.
Scientists create compounds called correctors to fix the CFTR proteins so that proper amounts transit to the cell surface where they can serve as chloride channels to help maintain a well-hydrated airway. Scientists also create compounds called potentiators to activate the CFTR channels at the cell surface to maintain the proper balance of electrolytes in fluids, including those found inside lungs.
The Science Translational Medicine study researchers note that Cystic fibrosis (CF) is caused by mutations in the CF transmembrane conductance regulator (CFTR), and that newly developed correctors such as lumacaftor (VX-809) that improve CFTR maturation and trafficking and potentiators such as ivacaftor (Vertex Pharmaceuticals trade name Kalydeco, developed as VX-770) that enhance channel activity have proven very effective for the three to five percent of CF patients who have a specific genetic mutation (G551D) that allows epithelial cells to transit enough CFTR proteins to the cell surface, but the chloride channels have a defective activation.
However, the scientists observe that for the vast majority of CF patients with the more common CFTR mutation F508 and insufficient CFTR proteins at the cell surface, a single compound is not sufficient treatment. Consequently, patients with F508 will likely require treatment with both correctors and potentiators to achieve clinical benefit, with the caveat that while acute treatment with this drug combination’s effectiveness of has been demonstrated in vitro, the researchers observe that impact of chronic therapy has not been established.
In studies of human primary airway epithelial cells, Dr. Gentzsch’s team found that both acute and chronic treatment with VX-770 improved CFTR function in cells with the G551D mutation, consistent with clinical studies. In contrast, chronic VX-770 administration caused a dose-dependent reversal of VX-809mediated CFTR correction in F508 homozygous cultures. This result reflected the destabilization of corrected F508 CFTR by VX-770, markedly increasing its turnover rate. The coauthors also note that chronic VX-770 treatment also reduced mature wild-type CFTR levels and function, and conclude that their findings demonstrate that chronic treatment with CFTR potentiators and correctors may have unexpected effects that cannot be predicted from short-term studies, and that combining these drugs to maximize rescue of F508 CFTR may require changes in dosing and/or development of new potentiator compounds that do not interfere with CFTR stability.
 “In our human airway epithelial model system, one of the drugs destabilizes and deactivates the protein that the other drug tries to correct,” explains Dr. Gentzsch, an assistant professor of cell biology and physiology, and senior author of the UNC Science Translational Medicine paper, in a UNC release. “Our data suggest that drug developers should take this destabilization effect into account in order to alleviate CF symptoms to a greater degree.”
“In our human airway epithelial model system, one of the drugs destabilizes and deactivates the protein that the other drug tries to correct,” explains Dr. Gentzsch, an assistant professor of cell biology and physiology, and senior author of the UNC Science Translational Medicine paper, in a UNC release. “Our data suggest that drug developers should take this destabilization effect into account in order to alleviate CF symptoms to a greater degree.”
A major focus of Dr. Gentzsch’s research at the UNC Marsico Lung Institute/Cystic Fibrosis Research Center is biosynthetic processing and intracellular trafficking of CFTR. This multi-domain protein functions as a chloride channel in apical membranes of epithelial cells and in addition regulates other ion channels and transporters. More than 1500 different mutations in the CFTR gene have been identified in cystic fibrosis patients, however, a deletion of phenylalanine 508 (DF508) is most common and present in more than 90% of cystic fibrosis patients. The misfolded protein DF508 cannot mature conformationally and is recognized by ER quality control and therefore is not able to proceed to Golgi and plasma membrane. Growth of cells at reduced temperature or other manipulations enable the nascent mutant protein to avoid ER quality control and reach the cell surface, but it is rapidly cleared from the distal secretory pathway and degraded in lysosomes. Rescue of DF508 CFTR from ER retention is currently being intensively pursued as a potential therapeutic approach to alleviate cystic fibrosis disease. The Grentzsch lab has a specific interest in the endocytic trafficking of rescued DF508 CFTR and we have recently found that rescue of the mutant protein from ER retention by some means affects endocytic routing of other components e.g. cholesterol and glycosphingolipids.
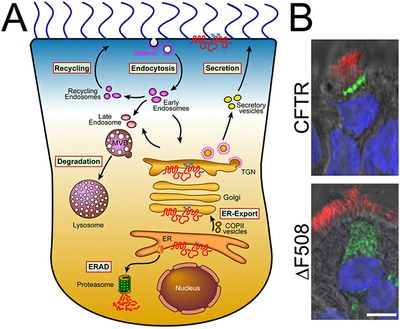
Image Caption: Intracellular trafficking of CFTR. A. Cartoon depicting intracellular CFTR trafficking routes. B. DeltaF508 CFTR has a processing and trafficking defect. In differentiated primary human airway epithelial cells, adenovirally overexpressed wild-type CFTR localized to the apical membrane, while deltaF508 was present only intracellularly, as shown by confocal microscopy of CFTR fluorescent labeling (bar=10µM; CFTR is shown in green, cilia that were labeled with anti-tubulin antibody in red and nuclei in blue) – Image Credit: Grentzsch Lab, UNC Marsico Lung Institute/Cystic Fibrosis Research Center
The most common mutation of CFTR, ∆F508, results in a misassembled protein that is retained at the Endoplasmic Reticulum (see above graphic) but can escape and proceed to the plasma membrane by addition of small-molecule correctors. However, rescued ∆F508 disappears rapidly from the cell surface and is subjected to lysosomal degradation, while wild-type CFTR is recycled back to the plasma membrane. The goal of research in Dr. Grentzsch’s lab is to elucidate a means to restore proper localization and stability of ∆F508 CFTR in human airway epithelia. Specifically, the researchers aim to identify therapeutic targets and pharmacological treatments that allow modulation of the cell surface density and stability of mutant CFTR proteins in cystic fibrosis.
In cystic fibrosis bronchial epithelia, deficient CFTR channel activity and the absence of CFTR-dependent inhibition of the epithelial sodium channel (ENaC) lead to dehydration of airway surfaces. While it has been established that mutations in CFTR result in defective regulation of ENaC and consequent sodium hyperabsorption in cystic fibrosis airways, the detailed mechanism by which CFTR mediates inhibition of ENaC remains unclear. ENaC is known to be stimulated by limited proteolysis of the extracellular domains and we have demonstrated recently that CFTR impedes proteolytic processing of ENaC.
In Dr. Gentzsch’s study, UNC postdoctoral fellow Deborah Cholon, PhD, conducted lab experiments using tissue samples from CF patients who have the most common CFTR transit genetic mutation (F508). The research team grew cultured CF epithelial cells in an environment that mimicked a human lung. Then the researchers treated the cells for two days with a corrector compound called VX-809, generic name lumacaftor. They observed that the amount of CFTR protein transiting the cell surface appropriately increased. But when they acutely (in seconds) added the potentiator VX-770, the corrected CFTR protein exhibited a brief increase in function that rapidly waned, a sign that the corrected CFTR protein was losing its ability to function as an ion channel.
In a separate experiment, Dr. Gentzsch’s team treated the CF cells for two days with a potentiator and a corrector at the same time; this is how a drug would be given to patients. Dr. Gentzsch found that the potentiator compound chronically destabilized the CFTR protein, and that the destabilization was dependent upon the dose of VX-770. That is, the experiments showed that the potentiator cancelled out the intended effect of having a corrected CFTR protein at the epithelial surface.
“The result was striking,” Dr. Gentzsch observes. “The potentiator acted like an inhibitor of the corrector compound. We could see the corrected CFTR protein disappearing.”
 The findings come one month after results from another clinical trial — unaffiliated with UNC — showed that the two-drug approach increased lung function in CF patients by 2.2 to 3.6 percent. This result was statistically significant, but Charles Esther, MD, PhD, a clinical associate professor whose Dr. Esther’s research interests concentrate on airway biomarkers, exhaled breath condensate, nontuberculous mycobacteria and respiratory infections in patients with CF, and one of the UNC coauthors, says that it would be difficult for many CF patients to perceive this level of improved lung function on a day-to-day basis. However, Dr. Esther and the UNC coauthors, who were not involved in the clinical trial, said that a statistically significant increase in lung function is thought to lead to fewer CF-related flare-ups over the course of a year.
The findings come one month after results from another clinical trial — unaffiliated with UNC — showed that the two-drug approach increased lung function in CF patients by 2.2 to 3.6 percent. This result was statistically significant, but Charles Esther, MD, PhD, a clinical associate professor whose Dr. Esther’s research interests concentrate on airway biomarkers, exhaled breath condensate, nontuberculous mycobacteria and respiratory infections in patients with CF, and one of the UNC coauthors, says that it would be difficult for many CF patients to perceive this level of improved lung function on a day-to-day basis. However, Dr. Esther and the UNC coauthors, who were not involved in the clinical trial, said that a statistically significant increase in lung function is thought to lead to fewer CF-related flare-ups over the course of a year.
Data from the clinical trial did show that the combination therapy substantially decreased the number of pulmonary exacerbations and hospitalizations, while also beneficially increasing the weight of CF patients. Drs. Gentzsch, Esther, and the UNC coauthors of the Science Translational Medicine paper agree that these clinical outcomes are promising and set the stage for new therapies for CF patients.
Dr. Gentzsch cites potential reasons to explain why patients in the clinical trial saw benefit from the two drugs even though her teams lab experiments produced evidence to the contrary. First, the destabilizing effects of VX-770 on the corrected CFTR protein might be less robust in the human body than were the effects seen in lab tests using human lung cells. In addition, cell culture studies lasted two days in her study, whereas the clinical studies lasted for months.
Dr. Gentzsch also says that the drugs may produce beneficial effects by mechanisms unrelated to correcting the mutant CFTR protein. For example, her research team found that VX-770 can decrease the function of a sodium channel in CF epithelial cells and other cells. Researchers also have found that VX-770 has antibacterial properties.
“The trial is important for gene research and may be a proof of principle that targeting the most common mutation in cystic fibrosis is an effective approach for treatment of most patients,” Dr.Gentzsch says. “But evidence of corrective effects on CFTR in the clinic requires further study. And we think that many people with CF need therapies that improve their health to a greater degree. We hope that our studies will help in this endeavor.”
This research was funded by the National Institutes of Health, the Cystic Fibrosis Foundation, and the Else Krner-Fresenius-Stiftung.
Sources:
UNC School of Medicine
Science Translational Medicine
Image Credits:
University of North Carolina



