Cystic Fibrosis Mutation May Change How Proteins Communicate



Protein interactions could be misdirected in people with cystic fibrosis (CF), causing a loss of normal protein function, according to researchers from the Scripps Research Institute (TSRI) in San Diego, California. The new study, titled “∆F508 CFTR interactome remodelling promotes rescue of cystic fibrosis“ appeared online on Nov. 30, 2015, ahead of its print publication in the journal Nature.
CF is one of the most common chronic lung diseases of children and young adults, and can be a life-threatening disorder. Breathing is often difficult for people with cystic fibrosis, due to a sticky mucus that builds up in the lungs and causes serious bacterial lung infections.
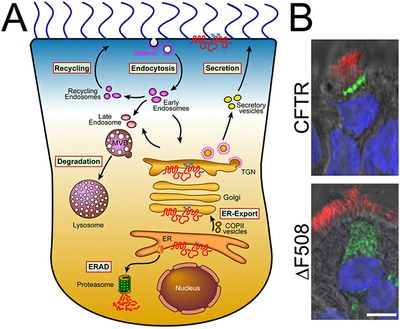
Mutations in the CFTR gene are the major cause of this mucus build-up. CFTR, or Cystic Fibrosis Transmembrane Conductance Regulator, helps shuttle chloride ions in and out of cells, balancing salt and water in the cells that line the lungs. Most people with cystic fibrosis have a ΔF508 mutation in the gene that encodes CFTR.
In the current study, scientists used a tool known as Co-Purifying Protein Identification Technology (CoPIT) to identify proteins in biological samples from people with CF. CoPIT allowed them to visualize CFTR protein partners and look for “incorrect protein cross-talk.” Surprisingly, when the CFTR protein contained the common ΔF508 mutation, it interacted with a set of completely different proteins from that of the normal, functional form of CFTR.
According to TSRI Staff Scientist and study co-author Sandra Pankow, “Three hundred proteins changed their level of interaction, and an additional 200 proteins interacted with the mutated CFTR. It’s like the wrong people are talking to the mutated CFTR all the time.” Better understanding of these “incorrect” protein interactions could lead to promising new ideas in CF treatments.
“The proteins and the interactions we’ve identified really fuel the pipeline for new drug targets to treat cystic fibrosis,” said Casimir Bamberger, a research associate in the lab of TSRI Professor John R. Yates and a study co-author.
The researchers were able to identify eight key proteins responsible for the incorrect interactions. Using genetic techniques to block these proteins appeared to restore normal CFTR function. The eight candidates may well be targets for future CF drugs or gene therapies.