
#NACFC2016 – Getting the Upper Hand on Mucus in Cystic Fibrosis
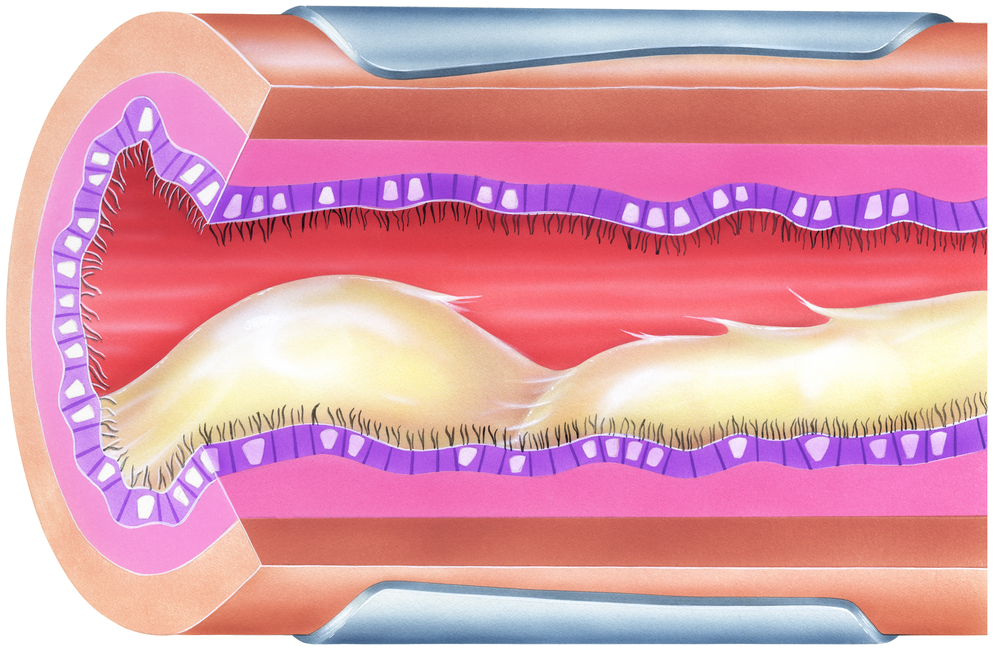



Abnormal mucus clearance in patients with cystic fibrosis (CF) may be improved with specific treatments, but despite intense research on the subject, there are many unanswered questions about how atypical mucus contributes to the disease and how to best improve the problem.
Researchers debated the issue in three discussions at a symposium titled “Mucociliary Transport and Ion Flux: Lessons Learned From New Models and New Drugs,” today at the 30th Annual North American Cystic Fibrosis Conference (NACFC) in Orlando.
An effective production of mucus and its clearance is needed to keep airways free of dirt and infectious agents. Researchers agree that for normal processes to work best, several factors are required.
Molecules called mucins can be considered the backbone of mucus, but a sufficient amount of liquid is also needed to prevent the mucus from becoming too thick. The beating of airway cilia — tiny appendages that cover the airway surfaces like short fur — is crucial for transporting mucus out.
Two types of mucins are produced in the airways, MUC5AC and MUC5B. They are synthesized in epithelial cells that line the airways and in glands. Studies have shown that CF patients have increased numbers of glands and produce more of the MUC5B mucin.
The production of the factor is increased during certain infections and in inflammatory conditions, such as asthma. Levels of the factor correlate with the capacity of mucus clearance.
Researchers know that insufficiently low levels of liquid in mucus caused by CFTR mutation makes mucus thick and more likely to plug the airways. Burton Dickey, MD, from the University of Texas MD Anderson Cancer Center, pointed to mounting evidence that a significant high release of mucins make mucus stick to the airway walls and obstruct the passage of air.
Animal experiments show that increased production of mucins, particularly MUC5AC, causes plugging of the airways. The mechanisms have been studied more in asthma than in cystic fibrosis, but Dickey believes they may also be important in CF patients.
Studies showed that the release of mucins occur at either a background level or at an increased rate that can exceed the background level by more than a thousand times when airways are infected or inflamed.
Because background levels and triggered mucin production are guarded by distinct molecular events, Dickey argued that drugs could be used to reduce the triggered rate without affecting background mucin levels.
In addition to such specific drugs, studies show that aerosols containing 7 percent saline may improve mucus clearance by depleting cellular stored mucins.
Steven Rowe, MD, from the University of Alabama, emphasized that in addition to the role of mucins and a lack of liquid making mucus too thick, there is evidence that other abnormalities of the composition of mucus may contribute to the clearance problem.
DNA released from dying bacteria, epithelial cells, and inflammatory cells contribute to making mucus abnormally thick. With these findings in mind, Rowe maintained that aerosols containing DNA-cutting enzymes could be used to improve mucus clearance.
Scientists know that a mutant CFTR protein leads to a lack of chloride and bicarbonate release, which prevents the secretion of fluid. An abnormal sodium transport through a channel called ENaC is believed to further worsen the dehydration of mucus. The dry and viscous mucus prevents cilia from beating.
Despite all this knowledge, Rowe holds that researchers are currently not able to determine how much the various mechanisms contribute to disease in CF.
Studies are currently ongoing to explore whether blocking the sodium-mediated movement of fluid back into the tissue may improve mucus clearance independent of other CF disease mechanisms.
Treatments targeting abnormal mucus in CF are under development, according to Rowe, who mentioned saline aerosols and dry powder mannitol. Also, agents reducing mucus oxidation can make it less viscous.
Finally, Marcus Mall, MD, from the University of Heidelberg, focused his talk on the role of dehydration in mucus plugging and reduced clearance. Studies in mice show that dehydration of airway surfaces is enough to cause an abnormal clearance of mucus, even when mucus production is not increased.
Other mouse experiments showed that when mucus production was increased in mice, liquid production was ramped up as well. When researchers prevented liquid production in the mice, mucus started plugging the airways, indicating that it is the liquid component that determines if mucus gets stuck in the lungs.
Studies also showed that mucus plugs trigger inflammation in the airways (even without an infection present) and dry mucus compresses cilia, preventing them from beating properly.
Mall noted saline as a means of improving mucus clearance, although he focused on the hydrating aspect of the treatment. Other approaches to improve airway surface liquid secretion could involve CFTR modulating drugs or activation of other chloride channels (for example, through the compounds TMEM16A and SLC26A9).
Drugs acting on the sodium transporter ENaC could be used to increase the levels of airways surface liquid, improve mucus clearance, and reduce plugging in cystic fibrosis patients.
Measuring mucus concentration and clearance will also be critical in determining the effectiveness of new treatments, Mall emphasized. In addition, these measurements will be needed to reach a better understanding of the relationship between airway surface moisture, inability of cilia to do its job, chronic inflammation, and infection in cystic fibrosis.



