
The Epithelium Sodium Channel and Cystic Fibrosis
Written by |





Cystic fibrosis is a complicated disease. As patients, we are taught that mutations in our cystic fibrosis transmembrane conductance regulator (CFTR) proteins are what lead to many of our symptoms and disease trajectories. However, research has shed light on many other proteins and biological processes that also dictate the course of our disease.
The epithelial sodium channel (ENaC) is an example of another ion channel shown to play a role in cystic fibrosis. Below, I explain the role of ENaC in cystic fibrosis, and also discuss a potential treatment strategy designed to help everyone with cystic fibrosis regardless of their CFTR mutation.
It’s imperative that we research treatments for all patients with CF, not just for those of us who have CFTR modulator medicines available. CF genotypes are complicated, and many mutations will not have a therapy available in the near future.
The ENaC protein
The CFTR and the ENaC are ion channels that reside on the surface of cells in the lungs. Ion channels move ions such as sodium and chloride across cells. Additionally, these channels regulate the amount of fluid in the lung. This fluid is critical to keeping mucus hydrated and freely moving, which is why those with CF often inhale a cocktail of medications to allow the mucus to clear from the lungs.
The CFTR, which helps add fluid to the surface of the lung, does not work in those of us with CF, while ENaC, which removes fluid from the surface of the lung, is working too much. This results in the dehydration of mucus and allows it to collect in the lungs. This accumulation of dehydrated mucus provides the ideal environment to support the development of infections that many of us battle (Figure 1).
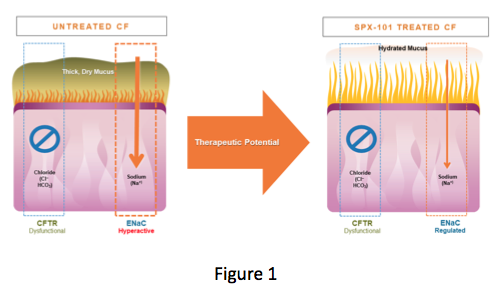
(Courtesy of Spyryx Biosciences)
Because ENaC is involved in mucus hydration independent of a CF mutation, scientists and clinicians have long sought to develop therapies to inhibit its activity. The idea has always been that inhibition of ENaC would improve mucus hydration and reduce lung infection by preventing the buildup of dehydrated mucus.
Because ENaC function is independent of CFTR mutation, such a therapy could be used by all CF patients. Early attempts to inhibit ENaC were limited by the short duration of action of these therapies and, in some cases, toxicity.
It’s important to remember that the fight to cure CF will occur on multiple fronts. For those of us who are adults with CF, we can’t expect that CFTR modulators alone will help overcome the damage accumulated over years of infection. This is why the following study excited me: If successful, it could serve as a new way to address another avenue that causes our mucus to become thick.
SPX-101’s mechanism
Spyryx Biosciences has developed SPX-101, an inhaled peptide intended to work differently than previous ENaC-target therapies. Rather than blocking the channel like a plug in a drain, SPX-101 aims to remove ENaC from the surface of airway cells. Because the channels have been removed, ENaC activity would be reduced and airway hydration would be restored, if successful. As a result, mucus clearance from the airways would be improved.
This mechanism showed promise in animal models and is now being tested in clinical trials. In June 2017, Spyryx presented data from a Phase 1 clinical trial (NCT03056989) that “demonstrated the clinical safety and tolerability of SPX-101 for up to 14 days in healthy adults ages 18-50 with no history of respiratory disease and with normal lung function,” according to a company news release.
A Phase 2 trial called HOPE-1 (NCT03229252) is currently enrolling participants in Canada, the United Kingdom, France, and Portugal. The 28-day placebo-controlled study will further test SPX-101’s safety and effectiveness in treating CF. More information, including study locations and contacts, can be found here.
SPX-101 studies
Following are some of the models that Spyryx used to indicate that SPX-101 has the potential to increase mucus movement.
First, Spyryx tested the ability of SPX-101 to increase mucus movement in a CF mouse model. The results were published in a study titled, “SPX-101 Is a Novel Epithelial Sodium Channel–targeted Therapeutic for Cystic Fibrosis That Restores Mucus Transport,” in the American Journal of Respiratory and Critical Care Medicine. (Note: All investigational therapies that enter clinical trial start with animal models before making it to patients like us. Also, in this study, several authors reported being affiliated with Spyryx.)
By comparing the first two bars in Figure 2, you can see that the CF mouse model has less mucus movement than a wild-type animal (no CF). When treated with a single dose of SPX-101, mucus movement in the CF mouse model was seen to be improved — almost back to the level of a mouse without CF. Next, Spyryx used a model where mucus movement was detected in living sheep. Here, the potential power of SPX-101 over traditional ENaC inhibitors could be seen.
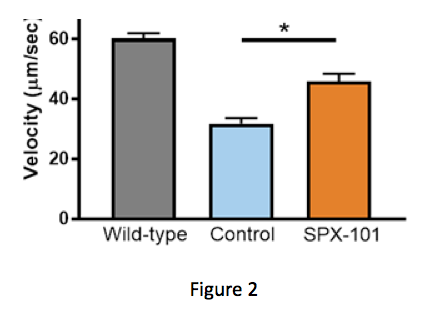
(Courtesy of Spyryx Biosciences)
In Figure 3 below, the graph shows the speed in which the mucus is moving over time. At the start of the experiment, the animals are treated with an inhibitor of CFTR. Addition of this inhibitor blocks CFTR function and reduces mucus velocity similar to what is observed in CF.
Four hours after addition of the CFTR inhibitor, animals were treated with either saline, amiloride (a classic ENaC inhibitor), or SPX-101. Both SPX-101 and amiloride were seen to significantly increase mucus velocity as compared to control immediately after treatment, according to the study. But while both treatments were seen to improve mucus velocity, only SPX-101 appeared to produce sustained recovery of velocity.
As the study progressed, the increase in mucus velocity associated with SPX-101 was maintained, the authors noted. In contrast, the positive effects of amiloride were not sustained and mucus movement fell back to diseased levels by the end of the study.
This could potentially impact those of us with CF. For starters, sustained activity over time could lead to fewer treatment doses and improved mucus clearance throughout the day. For many of us, we often feel worse by the time we have to do our next treatment. This is because the effects of our last airway clearance have worn off. The possibility of sustained mucus clearance would be an exciting possibility.
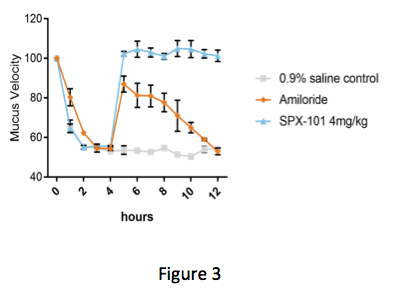
(Courtesy of Spyryx Biosciences)
In my opinion, CF will take a combination of multiple medications to truly restore and maintain healthy lung function. Unfortunately, some mutations will not have medications available to them in the near future. This is why those of us in the CF community need to advocate especially hard for members with rare mutations. CF will not be cured until all mutations are addressed.
***
Note: Cystic Fibrosis News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. The opinions expressed in this column are not those of Cystic Fibrosis News Today, or its parent company, Bionews Services, and are intended to spark discussion about issues pertaining to cystic fibrosis.



Leave a comment
Fill in the required fields to post. Your email address will not be published.