Fact-checked by
Fact-checked by 
Cystic fibrosis treatment
There is no cure for cystic fibrosis (CF). However, a variety of therapies are available to help with symptom management and to deal with complications of the genetic disorder.
CF treatments can slow disease progression and help patients maximize their quality of life.
The inherited disorder is characterized by the production of abnormally thick, sticky mucus. This mucus builds up in the lungs and other organs, interfering with their function and causing progressive damage in the long term. This thick mucus is ultimately responsible for most symptoms of CF, but patients can use available treatments to try to minimize their impact on daily living.
CFTR modulators
CFTR modulators are a recently developed class of medications that can help to boost the functionality of the protein whose impairment causes CF.
The disorder is caused by mutations in the gene that provides instructions for making the CFTR protein. This protein normally sits at the surface of cells and acts like a gated channel, regulating how water and salt molecules flow in and out of the cells. In CF, the protein does not function properly, which disrupts this flow and ultimately leads to the production of the thick, sticky mucus.
CFTR modulators are designed to improve the function of CFTR in people with specific disease-causing CFTR gene mutations. By improving the protein’s functionality, CFTR modulators can help people with CF to produce more normal, slippery mucus, which can ease system symptoms and slow disease progression.
There are two types of CFTR modulators, known as potentiators and correctors.
Potentiators
CFTR potentiators are molecules that can help to essentially prop open the gate-like CFTR protein on the cell surface. On their own, these therapies may be effective in people with mutations where the CFTR protein is able to get to the cell’s surface, but isn’t fully functional once there.
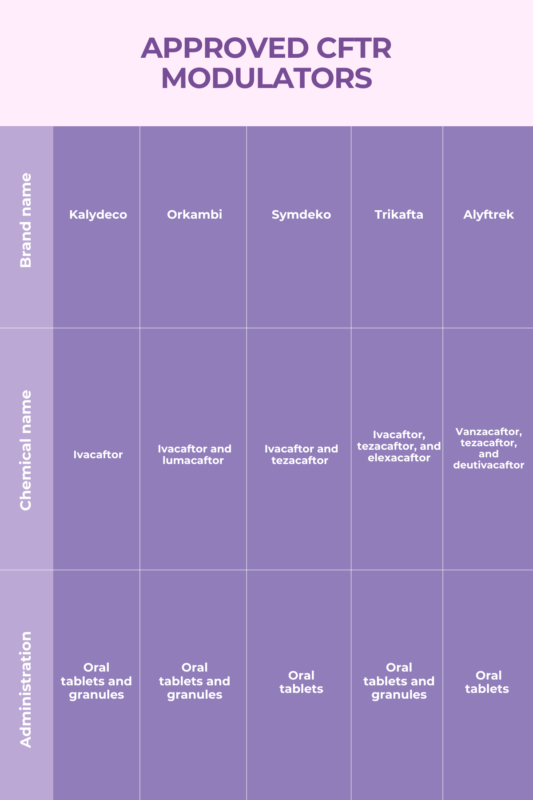
The oral potentiator therapy Kalydeco (ivacaftor), sold by Vertex Pharmaceuticals, is approved in the U.S. to treat CF patients as young as 1 month old who have one of 97 genetic mutations.
Correctors
Some CF-causing mutations — including the most common mutation, called F508del — make the CFTR protein unstable, so it gets destroyed before it can get to the cell’s surface. Correctors are molecules that can stabilize the protein, helping it reach the surface of the cell where it is needed to function.
There are four widely approved therapies that contain CFTR correctors. They work alongside potentiators that help the protein to function once the correctors have done their work and the protein is at the cell surface. They are:
- Orkambi (ivacaftor/lumacaftor)
- Symdeko (ivacaftor/tezacaftor)
- Trikafta (ivacaftor/tezacaftor/elexacaftor), sold as Kaftrio in Europe
- Alyftrek (vanzacaftor/tezacaftor/deutivacaftor).
In the U.S., Orkambi is approved for patients 1 year and older who have two copies of the F508del mutation. Symdeko is approved for patients ages 6 and older, whereas Trikafta is for those who are 2 and older. Alyftrek, the most recent of these therapies, is for patients 6 and older. Each treatment is approved for patients who have F508del or other CF mutations that are responsive to therapy. All four of these CF medications are sold by Vertex.
Antibiotics
The thick mucus that builds up in the lungs of people with CF makes them prone to bacterial infections. Antibiotics are prescribed to prevent, eliminate, or control respiratory infections caused by bacteria in CF patients.
By stopping or preventing damage caused by lung infections, antibiotics help to maintain lung function and quality of life in CF patients. Such cystic fibrosis treatment also reduces exacerbations — periods when lung function suddenly declines, usually due to a lung infection.
There is a complex arsenal of antibiotics for CF. Some antibiotics are only effective against certain bacteria. Others, called broad-spectrum antibiotics, attack a wide range of bacteria. Identifying the right combination of antibiotics for a patient early on can help prevent the infection from spreading.
Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa, and Burkholderia cepacia are some common bacterial species that often infect CF patients. Most commonly used antibiotics in CF target one or more of these species.
Oral antibiotics
Oral antibiotics may be used to help manage chronic infections or mild exacerbations. These therapies are available as liquids, tablets, or capsules. Some are administered once daily for a few days, while others can be used for longer periods. Some can be taken with food, while others should be taken on an empty stomach.
It is important to read the prescribing clinicians directions to ensure these medications are taken as directed.
Oral antibiotics that are often used in CF include:
- azithromycin, sold as Zithromax and Zmax, among others
- cephalosporins, a group of antibiotics that includes cephalexin, cefdinir, cefprozil, and cefaclor
- penicillins
- quinolones such as ciprofloxacin or levofloxacin
- sulfonamides (also called sulfa drugs), particularly sulfisoxazole, sometimes given in combination with erythromycin
- tetracyclines including tetracycline, doxycycline, and minocycline
- linezolid, sold as Zyvox and generics.
Inhaled antibiotics
Inhaled antibiotics are delivered directly into the lungs, typically via the use of a nebulizer that creates a mist of therapy that a patient can breathe in. These therapies often are used to help manage chronic infections and reduce the need for into-the-vein (intravenous, or IV) antibiotics.
CF specialists usually determine if a patient needs to take an inhaled antibiotic based on the results of a sputum culture that identifies a specific type of bacterial infection.
Aztreonam and tobramycin are inhaled antibiotics approved to treat CF patients with P. aeruginosa infections. An inhaled form of amikacin, called Arikayce, has been approved for treating infections caused by Mycobacterium avium complex.
Patients usually take one of these antibiotics every other month for 28 days. However, doctors may have patients alternate between antibiotics for the best effect.
To allow the antibiotic to reach deep into the lungs, inhaled antibiotics should be taken after other treatments that clear mucus as much as possible, like bronchodilators, mucus thinners, and airway clearance techniques.
IV antibiotics
Intravenous (IV or into-the-vein) treatment is often required for serious pulmonary exacerbations in CF patients in addition to inhaled or oral antibiotics.
IV antibiotics are delivered directly into the bloodstream, usually through a catheter placed in the arm. These antibiotics are typically given by healthcare professionals in the hospital so that the dosage and potential side effects can be carefully managed, though sometimes the treatment can be self-administered at home after proper training.
Among the IV antibiotics that may be used for infections in CF patients are:
- gentamicin
- amikacin
- tetracyclines
- penicillins
- quinolones
- linezolid.
Inhaled therapies
CF typically has a substantial impact on the lungs, resulting in symptoms like coughing, wheezing, and shortness of breath. Inhaled therapies, administered directly into the lungs, may be used to help ease these symptoms.
Some inhaled therapies are administered via metered-dose inhalers, which provide a set amount of the therapy via a spray. Others are administered via nebulizers, which use systems like compression or vibration to turn a liquid medication into a mist that can be inhaled.
Bronchodilators
Bronchodilators are medications that cause the airways to relax and widen, making it easier to clear mucus and allowing the lungs to take in more air. These therapies are commonly prescribed to help manage CF lung symptoms. Bronchodilators also can be taken before other inhaled medications to open the airways, allowing the medicine to penetrate deeper into the lungs.
Bronchodilators that are often used in CF include:
- albuterol (also known as salbutamol), sold as Ventolin, ProAir, and Proventil, among others
- levalbuterol, sold as Xopenex and generics
- Combivent (albuterol plus ipratropium bromide).
Inhaled corticosteroids
Corticosteroids, also referred to as steroids, are anti-inflammatory medications. They may be used to help with cystic fibrosis symptom management in wheezing or chest tightness in patients who are continuing to have bothersome symptoms despite the use of bronchodilators. It’s generally recommended that a corticosteroid should be considered if someone needs a bronchodilator several times per week.
There are several commonly used inhaled corticosteroids in CF, including:
- fluticasone, sold as Flovent and Flonase among others
- beclomethasone, sold under brand names such as Beclovent, Vanceril, and QVAR
- budesonide, such as Pulmicort.
Mucolytics
Mucolytics, also called mucus thinners, are medicines that work to break up thick mucus, making it more watery so it’s easier to clear from the lungs.
Common mucolytics used in cystic fibrosis include:
- Hypertonic saline
- Pulmozyme (dornase alfa)
- Bronchitol (mannitol)
- acetylcysteine (also called N-acetyl cysteine or NAC), sold as Mucomyst, among others.
Nutrition-related treatments
In many people with CF, mucus builds up in the pancreas which prevents certain enzymes from getting into the digestive tract. This condition, called exocrine pancreatic insufficiency or EPI, makes it harder for the body to digest certain kinds of food, particularly fats.
PERT
Pancreatic enzyme replacement therapy, known as PERT, is a treatment for EPI that involves administering versions of the enzymes that are missing, which can improve patients’ ability to digest their food. This can help individuals with CF to get adequate nutrition, as well as aid in easing symptoms caused by poor digestion, such as foul-smelling stools or bloating.
Several PERT options, which typically use enzymes derived from pigs, are available. These include:
- Creon
- Pertzye
- Pancreaze
- Relizorb
- Viokace
- Zenpep.
Probiotics
Probiotics, which are supplements containing so-called good bacteria, are often used to help manage digestive issues in people with CF. These supplements can help to essentially repopulate the gut with bacteria following antibiotic treatment.
At present, there’s not much data on whether and how probiotics might help people with CF, and there’s a lot of variability among the data that are available — for example, some studies have suggested that probiotic supplements can help reduce the occurrence of pulmonary exacerbations in CF, but other research has found no such effect.
It’s also important to note that probiotics are supplements, not medications, which means that in the U.S. they are subject to much less stringent regulations. It is recommended that CF patients talk with their healthcare team before starting on any new probiotics or other supplements.
Other treatments
In addition to CFTR modulators, antibiotics, and other medications, a number of nondrug therapies are commonly used to help manage health and improve quality of life for people with CF.
Physiotherapy
In physiotherapy, also called physical therapy, a therapist works with a patient to perform exercises to support health. Physical therapy in cystic fibrosis often includes techniques that aim to support respiratory function and help clear mucus out of the lungs. Physical therapist usually give patients exercises to do at home to supplement their sessions.
Oxygen therapy
Oxygen therapy, in which a patient breathes oxygen through a mask or nose tube, allows patients to take in more oxygen than they normally would. This can help ease symptoms like breathlessness and fatigue, and it may ensure patients can get sufficient oxygen while sleeping or exercising.
Techniques and devices to clear mucus
People with CF often rely on airway clearance techniques to help them clear the thick mucus out of their lungs. These strategies generally involve coughing or huffing to get mucus up, along with breathing techniques to help dislodge mucus in the lungs. Patients are advised to work with their healthcare team to figure out what technique or combination of techniques will work best for them.
Some devices may be helpful for clearing mucus. Among these are High-Frequency Chest Wall Oscillation (HFCWO) systems, which use a vest that puts pressure on the chest to help force out mucus. With Positive Expiratory Pressure (PEP) systems, a patient breathes into a mouthpiece that gives resistance, forcing the person to exhale harder, which helps bring up mucus. Some PEP systems also include vibration to help dislodge mucus, known as oscillating PEP.
Creating a treatment plan
Managing cystic fibrosis is a lifelong process. It’s recommended patients and their families work with an interdisciplinary team of healthcare providers to come up with a treatment plan that’s right for them.
The CF care team generally includes doctors, nurses, and therapists covering a variety of specialties, from lung health to psychology. These clinicians can work with the patient to create an individualized treatment plan that covers all of the individual’s needs. In addition to prescribing cystic fibrosis medications, the treatment plan also usually involves guidance on nutrition and exercise.
This process generally begins soon after a person receives a cystic fibrosis diagnosis — which nowadays usually happens in infancy due to the widespread adoption of newborn screening programs. After positive screens, typically done within hours of a baby’s birth, a sweat chloride test is usually done to support a CF diagnosis. Genetic testing may be used to either confirm diagnosis or evaluate a patient’s potential eligibility for treatment with CFTR modulators.
Early CF treatment for children is tailored based on the child’s experience — therapies are given to help improve breathing, support digestion, and clear up infections as needed. As a person with CF grows up, it’s important to continue having regular conversations about continuing care. Patients should be included in these conversations as appropriate.
A good treatment plan will be continually adjusted and tailored to address the patient’s current situation. Importantly, some treatments — including many CFTR modulators — are only approved for use in children after a certain age. Thus, needed new therapies should be added to the treatment plan as the individual gets older.
As people with CF grow through adolescence into adulthood, most will gradually transition from a CF center that specifically treats children to one that specializes in adults. While familial support is always important, most adults with CF live independently and primarily manage their own care.
Cystic Fibrosis News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Your CF Community

Visit the Cystic Fibrosis News Today forums to connect with others in the CF community.
FAQs about cystic fibrosis treatment
Category:
Treatment
Although there isn’t a cure for the disease, numerous treatments are available to help ease symptoms of cystic fibrosis, including therapies to ease breathing, clear mucus, and improve digestive health. CFTR modulator therapies also may help to ease CF symptoms in patients with amenable mutations.
Category:
Treatment
Many treatments for cystic fibrosis are covered by insurers, but specific policies for individual medications will vary depending on the insurer. Patients are advised to talk with their insurance provider about coverage that’s available to them. According to the Cystic Fibrosis Foundation, Medicare is a coverage option for some patients.
Category:
Treatment
There is no cystic fibrosis (CF) cure. However, a variety of medications and other therapies are available that can help to manage CF, slow the disease’s progression, and improve quality of life for patients.
Category:
Treatment
While there is no cure for cystic fibrosis (CF), a number of therapies are available to help manage the disease. These include antibiotics to deal with recurrent infections, enzyme supplements to manage nutrition problems, and inhaled therapies to ease breathing. CFTR modulators, a class of medication that can ease symptoms and slow disease progression by acting directly on the protein whose mutation causes CF, are available for patients with specific disease-causing mutations.
Category:
Treatment
Managing cystic fibrosis (CF) usually involves combinations of different therapies to address the individual’s symptoms and limit future problems. It’s recommended that people with CF consult with a multidisciplinary healthcare team to come up with an individualized treatment plan based on their specific situation. Patients’ needs, and the effectiveness of given treatments, are likely to change as individuals get older.
Related Articles
-
-
 Discussion
Discussion
-
-
 Discussion
Discussion
-

-