
The Evolution of CF Treatments
Written by |




Cystic fibrosis care has seen such rapid advances that the average CF patient has likely seen a dramatic evolution in treatment strategies in their lifetime. Here are some of the biggest milestones that shaped modern-day CF treatments.
(Dates are when these therapies and ideas were adopted. Many are no longer relevant due to antibiotic resistance, new studies and philosophies, and creation of better therapies.)
Antibiotics
1940s
- Nebulized penicillin is used for chronic coughs.
- Sulphadiazine (sulfa) drugs are also used but later thought to be ineffective.
1950s
- The antibiotics chlortetracycline and oxytetracycline are the first-choices for treating infections. Stronger doses in combination with antibiotics chloramphenicol and erythromycin are used for more dangerous infections.
1980s
- IV antibiotics gentamicin and tobramycin are used widely for CF patients.
1990s
- The antibiotic flucloxacillin is used regularly. The Antibiotics Group of the UK CF Trust recommends the drug be given continuously for the first two years after birth for all CF patients.
- A special formulation of tobramycin (Tobi) is created for inhalation.
2000s
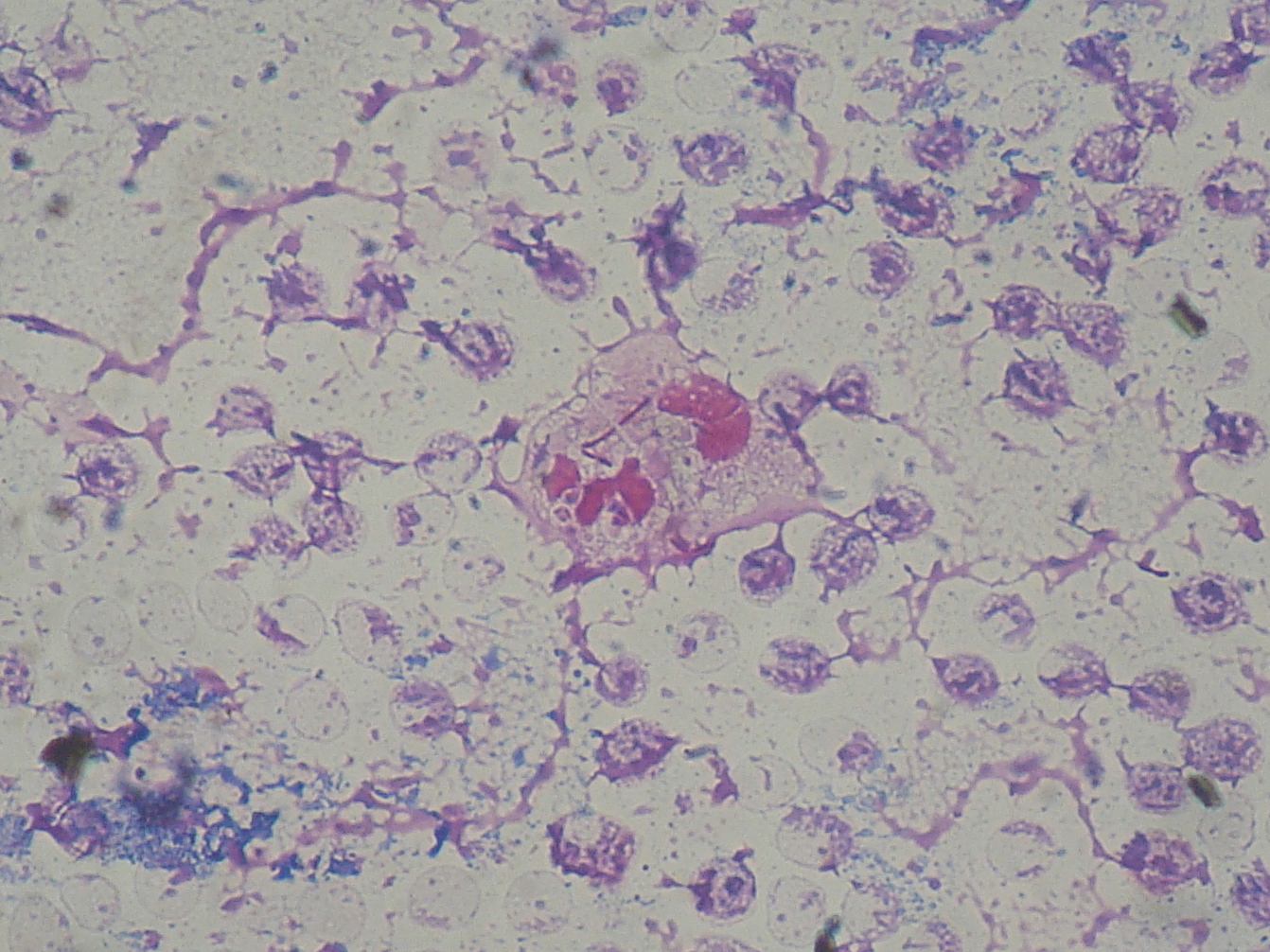
Pseudomonas aeruginosa (bastonete Gram negativo)
- Azithromycin is used for patients with chronic bacteria Pseudomonas aeruginosa and for those with severe inflammation.
- IV antibiotics are used more often in early stages of infections rather than as a last resort.
- An inhaled medicine called Cayston is created to fight Pseudomonas.
Today
- Antibiotic formulations such as Lynovex are being developed.
- New antibacterial treatments, such as IV gallium and inhaled nitric oxide, are being tested with much promise.
MORE: Why the Maui Ola Foundation is helping patients surf
Mucus thinners/physiotherapy
1950s
- Iodides, oral streptokinase or streptodornase, and intramuscular or inhaled pancreatic trypsin are used to thin CF mucus.
- Patients sleep in “mist tents” filled with misted 10 percent solution of propylene glycol and 3 percent saline. Mist tents are discontinued by most clinics in the 1970s.
- Clapping on the back and pressure vibrations are used as physiotherapy to remove mucus from CF lungs.
1960s
- N-acetylcysteine (NAC) is inhaled or taken orally to thin mucus.
1990s
- The benefits of exercise for removing mucus from lungs and increasing lung function is paid more attention.
- Recombinant human Dnase (Pulmozyme) thins mucus when inhaled. It is the first truly effective mucus thinner.
Nutrition
1940s
- Pathologist Dorothy Andersen advises for CF patients: “A low-fat, high-protein diet with a liberal allowance of vegetables, fruits and sugar and moderate restriction of starch. Supplementary vitamin A is essential and pancreatin and vitamin B complex are given.”
1950s
- Pancreatic enzymes are found to be effective in treating many CF patients’ inability to absorb nutrients.
1970s
- A high saturated fat diet of whole milk, butter, eggs, and animal fats results in weight gain for Canadian Dr. Crozier’s patients.
- Canadians CFers, who had superior nutritional statuses, live longer than other patients across much the world. This is still true.
- Dr. Allan of England popularizes a nutritional supplement that is made of beef serum protein hydrolysate, glucose polymer, and medium chain triglycerides. The “Allan Diet” is successful in weight gain. Supplements would soon be widely used for extra calories, fat, and protein in the CF diet.
- Acid-resistant enzymes, Creon and Pancrease, are released. They are more effective than predecessor Cotazym, which was a powder whose absorption was affected by stomach acid.
1980s
- Vitamin doses are better measured to account for fat-soluble deficiencies.
- Feeding tubes are used for patients who have extra difficulty in gaining weight.
MORE: Lung-themed watches sold to benefit CF research
Diagnosis
1938
- Dr. Dorothy Anderson creates the first description of CF based on autopsy findings of children dying of malnutrition, calling it “cystic fibrosis of the pancreas.”
1950s
- The sweat electrolyte defect in CF is discovered in 1953.
- Pulmonary function testing is used to diagnose and track CF patients. Historically, the taste of salty skin on a patient was a defining symptom of CF.
1960s
- Sweat tests (created in 1959) and biopsies of the small intestine are used to diagnose patients with CF.
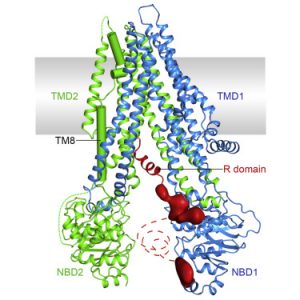
Molecular structure of the CFTR protein
1980s
- The CF Gene is identified in 1989. It is named the cystic fibrosis transmembrane conductance regulator (CFTR). The causes of CF are now better understood and the push for a cure gains momentum.
- The life expectancy for a CF patient is 12 in the United States and 20 in Canada.
2000s
- Full sequencing of the CF Gene, CFTR, is now available. Over 1,700 CF mutations are identified.
- The median life expectancy for CF patients is 47 as of November 2017.
Potential CFTR Correctors
1990s
- CF defects are corrected in the lab in 1990. The idea is applied to three mouse models with varying but disappointing results. The mouse models opened the door to further in vivo experimentation of CFTR function and gene transfers. Gene therapy is thought to be the answer for a cure.
2000s
-
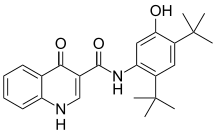
Kalydeco’s chemistry.
Pharma company Vertex releases Ivacaftor (Kalydeco) in 2012. It helps the CFTR function better for some mutations, decreasing major symptoms of CF and delaying the toll of the disease on lungs.
- Vertex combines Ivacaftor and a new medicine called lumacaftor to create Orkambi, released in 2015. Orkambi is meant to achieve the same results as Kalydeco for different mutations.
MORE: Four common misconceptions about cystic fibrosis
Cystic Fibrosis News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.




Leave a comment
Fill in the required fields to post. Your email address will not be published.